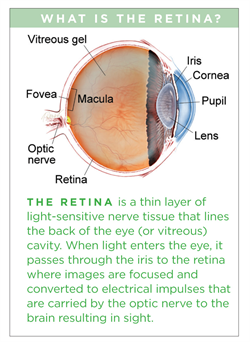
Retinoblastoma
is the most common primary tumor of the eye in infants and young children. This form of eye cancer is caused by uncontrolled division of the cells that make up the retina in the back of the eye, and requires prompt evaluation and treatment by a team of specialists.
Symptoms
• Leukocoria (white pupillary reflex)—In a flash photograph, the normal red reflex of the child’s eye(s) appears to be white, commonly described as a cat’s eye reflex.
• Strabismus (crossing of the eyes)—The child’s eye may appear to be drifting inward or outward, as a result of reduced vision in that eye from the tumor.
• Eye redness—The tumor can invade structures in the front of the eye that can cause glaucoma, inflammation, and abnormal appearance.
• Proptosis (sticking out of the eyeball)—If the tumor becomes large, it can result in the eye appearing to bulge out of the eye socket.
Causes
Retinoblastoma is caused by a mutation in the RB1 gene on chromosome 13. This gene is responsible for producing a protein that functions as a tumor suppressor, and every cell in the body has 2 copies of the gene. When both copies are mutated, the cell divides uncontrollably, leading to tumor formation.
About 60% of retinoblastomas are due to spontaneous (de novo) mutations that are nonhereditary. The remaining cases are due to a hereditary mutation that is passed down from one (or both) parents.
A mutation in the second copy of the gene then leads to tumor formation. Inherited retinoblastoma cases are more likely to involve both eyes with multiple tumors in each eye, and tend to develop before the first birthday, and may be associated with a separate tumor in the brain, called trilateral retinoblastoma in about 1 in 20 children with bilateral retinoblastoma.
Diagnostic testing
The first step is to perform a dilated fundus examination and carefully evaluate the retina and other structures of the eye. In children from 2 months to 5 years of age, this usually involves an examination under anesthesia. The following testing is usually performed:
- Fundus photography to document the appearance and size of the tumor in the eye and its response to treatment.
- Ultrasound of the eye to evaluate the size of the tumor, involvement of other structures in the eye, and the presence of fluid in the retina.
- MRI (magnetic resonance imaging) of the brain to look for tumors in the brain that can occur along with retinoblastoma. CT scans are usually avoided due to the increased risk of radiation exposure in children with retinoblastoma.
- Genetic testing to determine whether the child has an inherited mutation, which can guide treatment, monitoring, and screening of relatives and the child’s offspring.
Treatment and prognosis
Significant advancements have been made in retinoblastoma treatment since the 1990s to prevent spreading of the tumor and to preserve the eye.
- Systemic intravenous (through the vein) chemotherapy is the most common initial treatment, and usually involves a combination of 3 chemotherapeutic agents. Treatments are administered over multiple cycles spread over several months.
- Intra-arterial chemotherapy involves threading a thin tube called a cannula through the femoral artery in the thigh up to the blood supply of the brain and finally to the ophthalmic artery (behind the eye), where a chemotherapeutic drug (melphalan) is administered. This promising technique was developed in the mid-2000s and has since been increasingly refined.
-
Local intravitreal injection of chemotherapy directly into the eye is also used to deliver the treatment.
-
Laser treatment can be applied directly to smaller tumors (usually after receiving one of the forms of chemotherapy).
-
Enucleation, or removal of the eye, must sometimes be employed to prevent the spread of the tumor to the brain. This is done when the tumor is very large or does not respond to chemotherapy or laser treatment. The child is fitted with an artificial eye (ocular prosthesis).
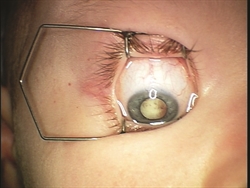
Figure 1: After placement of a speculum to hold the eye open during examination under general anesthesia, the tumor inside the patient’s eye is visible to the naked eye of the examiner. (Photo courtesy of G. Baker Hubbard, MD) |
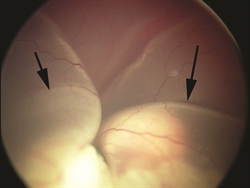
Figure 2: Ophthalmoscopy and photographs during examination reveal a large tumor with subretinal fluid that encompasses the eye. (Photo courtesy of G. Baker Hubbard, MD) |
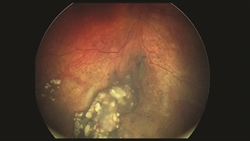
Figure 3: This is the patient’s eye after treatment with 2 rounds of intra-arterial chemotherapy, along with additional laser photocoagulation of the tumor. The tumor has responded well to treatment. (Photo courtesy of G. Baker Hubbard, MD) |
After treatment, close periodic monitoring is required to look for recurringtumors, including in the other eye in cases where only 1 eye is affected. The risk of recurrence is much lower after 7 years of age. Children with hereditary retinoblastoma also have an increased lifetime risk of cancer in other organs, including sarcomas of bone and soft tissue, melanoma (skin cancer), as well as brain, breast, and lung cancer.